使用 NCBI 在线 BLAST 进行序列比对
- 如何进入 NCBI BLAST 网站、界面长什么样
- 如何准备和粘贴示例序列
- 关键参数(数据库、算法、E-value 等)具体含义
- 如何阅读 BLAST 结果页面中的图形和表格
- 根据不同研究目的如何调整参数
一、BLAST 与 NCBI 在线平台简介
1.1 什么是 BLAST?
BLAST(Basic Local Alignment Search Tool) 是一类用于“在数据库中查找与给定序列相似序列”的局部比对算法家族,常见变体包括:
- BLASTN:核酸 vs 核酸
- BLASTP:蛋白 vs 蛋白
- BLASTX:核酸(翻译成蛋白)vs 蛋白
- TBLASTN:蛋白 vs 翻译后的核酸库
BLAST 的典型应用包括:
- 判断一个未知序列可能来自哪个物种、对应哪个基因或蛋白
- 查找同源基因(ortholog/paralog)
- 验证测序结果是否正确、是否被污染
1.2 NCBI 在线 BLAST 入口与界面
NCBI 提供了免费的在线 BLAST 服务,无需本地安装软件,只要有浏览器即可使用。
https://blast.ncbi.nlm.nih.gov/- Nucleotide BLAST(核酸 BLAST)
- Protein BLAST(蛋白 BLAST)
- BLAST Assembled Genomes 等高级入口
二、准备示例序列
为演示流程,先给出两条简短的示例序列,可直接复制使用。
2.1 核酸序列示例(nt BLAST)
下面是一段来自人类线粒体基因组(COI 片段)的核酸序列,已用 FASTA 格式 标注好:
>Example_COI_human_mtDNA
ATGATTATGGAAGCTTCTAGGAGCCATCAAGTATTTAGCTGACTATGAACCCCCT
ACAAATCATTAATCGGAGGCTTTGGCAACTGACTAGTTCCCCTAATAATCGGAGG
CTTTGGAAACTGACTAGTCCCTCTAATAATCGGAGGCTTTGGCAACTGACTAGTT说明:
- 第一行以
>开头的是序列 ID,可以任意命名 - 后面几行是碱基序列,只包含
A/C/G/T(以及少量其他 IUPAC 碱基符号)
2.2 蛋白序列示例(protein BLAST)
下面是一条简化的蛋白氨基酸序列,同样是 FASTA 格式:
>Example_Protein
MALWMRLLPLLALLALWGPGPGAGGHEGSLQPLALEGSLQKRGIVEQCCTSICSL
YQLENYCN说明:
- 第一行以
>开头的是蛋白 ID - 后面是氨基酸单字母码(A、R、N、D、...)
三、一步步完成核酸 BLAST(BLASTN)
3.1 打开 BLASTN 页面
- 访问
https://blast.ncbi.nlm.nih.gov/ - 在首页点击 Nucleotide BLAST 按钮,进入核酸 BLAST 页面。
进入页面后,可看到以下主要区域:
- Enter Query Sequence:粘贴或上传核酸序列
- Choose Search Set:选择比对的数据库和物种范围
- Program Selection:选择 BLAST 算法模式(Highly similar sequences / Somewhat similar sequences)
- Algorithm parameters:展开的高级参数区(Word size、Match/Mismatch scores 等)
3.2 粘贴序列与基本设置
在 Enter Query Sequence 区域进行如下操作:
- 将上文给出的核酸 FASTA 序列整体复制,粘贴到 “Enter Query Sequence” 的文本框中。
- 保持 Query subrange 为空(代表比对整条序列)。
- 如果有多个序列,可以继续往下粘贴,每条以
>开头。
在 Choose Search Set 区域:
- Database 建议初学者选择:nr/nt(Non-redundant nucleotide sequences)
- Organism:
- 如果已大致了解来源物种,可输入例如 “Homo sapiens (taxid:9606)”
- 如果完全未知,建议先留空,搜索所有物种
3.3 重要参数说明
在 Program Selection 和 Algorithm parameters 部分,有一些关键参数值得理解:
- Highly similar sequences (megablast):适合比对高度相似的序列,例如不同样本同一物种
- More dissimilar sequences (discontiguous megablast):适合更远缘的同源性搜索
- E-value 表示“在当前数据库大小下,随机出现同等或更好比对得分的期望次数”
- 数值越小,说明该比对越不可能是随机产生的,可信度越高
- 常见默认值为 10,对于高质量比对,可关注 E-value < 1e-5 或更小的 hits
- BLAST 首先寻找长度为 word size 的完全匹配“种子”
- word size 越小,敏感性越高,但速度更慢
- 对较短或较不保守的序列,可以适当减小 word size 提高灵敏度
- Match/Mismatch:匹配奖励与错配惩罚(如 +2/-3)
- Gap costs:插入/缺失的开启动作和延长动作的惩罚
- 默认设置通常已对大多数情况较为合理,一般无需修改
3.4 提交任务与运行过程
- 确认序列与参数设置无误后,点击页面下方的 BLAST 按钮。
- 页面会跳转到一个“搜索进行中”的等待界面,显示队列位置和预计剩余时间。
- 完成后会自动跳转到结果页面,包含多个标签页(Overview、Descriptions、Alignments、Graphics 等)。
四、一步步完成蛋白 BLAST(BLASTP)
4.1 打开 BLASTP 页面
- 同样访问
https://blast.ncbi.nlm.nih.gov/ - 在 BLAST 首页点击 Protein BLAST。
蛋白 BLAST 的页面结构与核酸 BLAST 非常类似,只是数据库和参数的默认设置稍有不同。
4.2 粘贴序列与参数设置
在 Enter Query Sequence 区域:
- 将上文给出的蛋白 FASTA 序列复制粘贴到文本框。
- 确认下方的 Query subrange 为空,表示使用全长蛋白。
在 Choose Search Set 中:
- Database:建议选择 nr(Non-redundant protein sequences)
- Organism:可留空,或填写预期物种(如
Homo sapiens)。
蛋白 BLAST 常见的额外参数:
- Matrix(替换矩阵):如 BLOSUM62、PAM30 等,决定不同氨基酸替换的得分
- Comp. based stats:成分校正设置,一般保持默认
设置好后同样点击 BLAST 等待结果。
五、结果页面结构与解读
无论是核酸 BLAST 还是蛋白 BLAST,结果页面结构非常类似,主要包含以下几个关键部分。
5.1 图形概览(Graphic Summary)
通常位于结果页面靠上的 “Graphic Summary” 区域,用彩色条带展示:
- 横轴:查询序列(Query),从 5' 到 3'(或 N 端到 C 端)
- 每一条彩色横线:一个数据库匹配(Hit)的局部比对位置
- 颜色深浅:代表得分高低(Score),颜色越深表示匹配质量越高
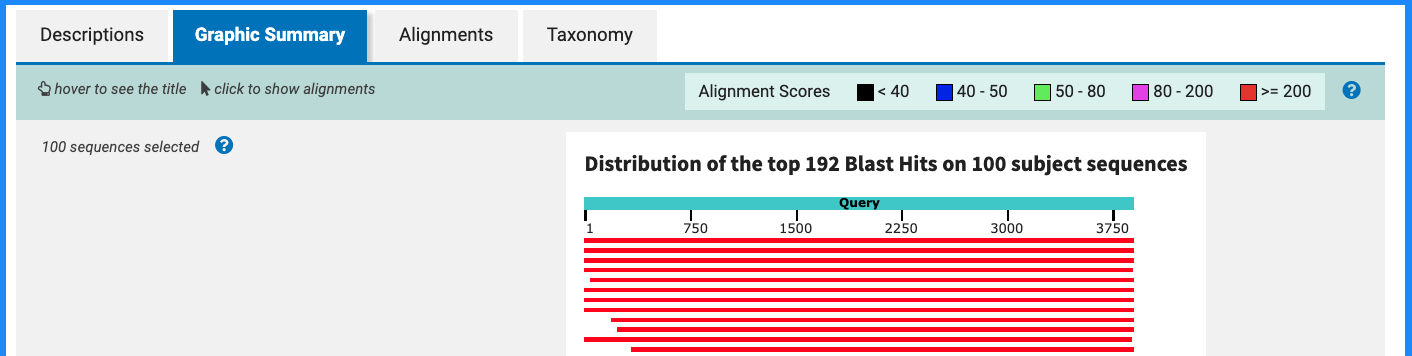
解读要点:
- 如果在整条 Query 上都能看到一条长而连续的深色条,说明有高度相似的同源序列存在
- 如果只在局部有短片段比对,可能对应保守结构域或局部保守区域
5.2 匹配列表(Descriptions)
在 “Descriptions” 或 “Sequences producing significant alignments” 表格中,常见列包括:
- Description:匹配序列的注释信息(基因名、物种、功能描述等)
- Max score / Total score:比对得分,数值越大表示比对越好
- Query cover:查询序列中被覆盖的比例(%)
- E value:显著性指标,数值越小越可信
- Per. Ident:身份性(Identity)比例,即完全匹配的碱基/氨基酸比例
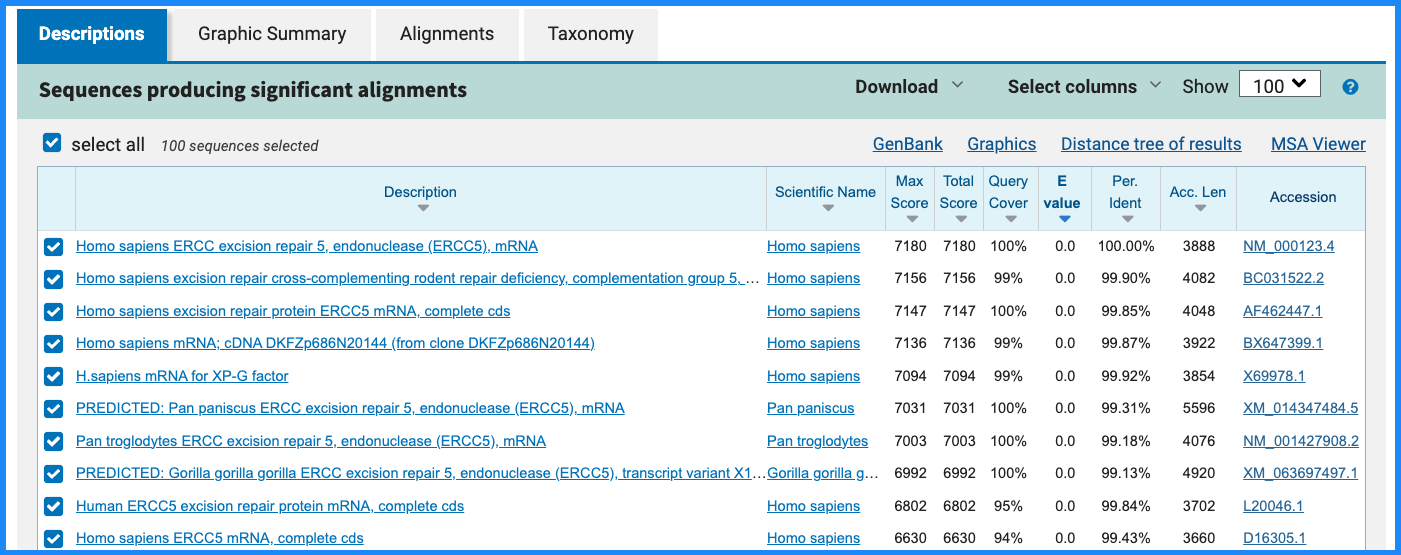
一般经验:
- E-value < 1e-5 且 Per. Ident > 80%,通常可以认为是比较可靠的同源关系(具体阈值依研究而定)
- Query cover 较低但 Identity 很高时,说明只是局部高度保守
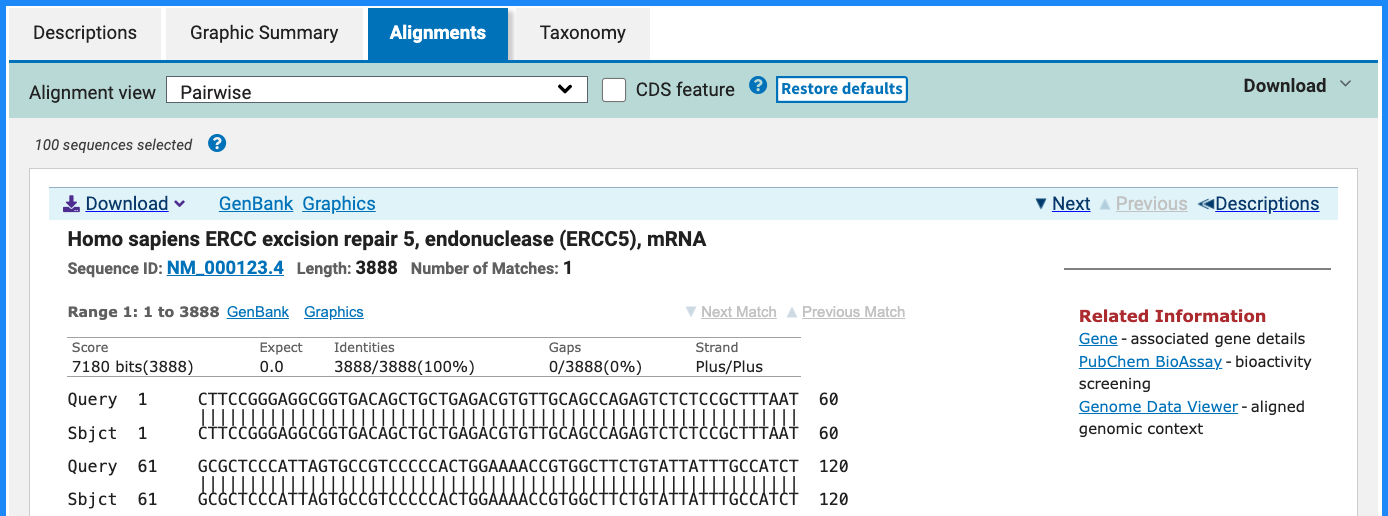
5.3 比对详情(Alignments)
点击某一条 Hit,可以看到具体的序列比对:
- Query:查询序列
- Subject:数据库中的匹配序列
- 中间一行的符号:
|:完全相同+:相近/保守替换(蛋白)- 空格或其他:错配或 gap
- Identities:完全匹配的碱基/氨基酸数量及比例
- Gaps:插入/缺失的数量
通过该区域可以直观判断:
- 是否存在大片段连续匹配
- 是否有大量 gap 或集中错配区域
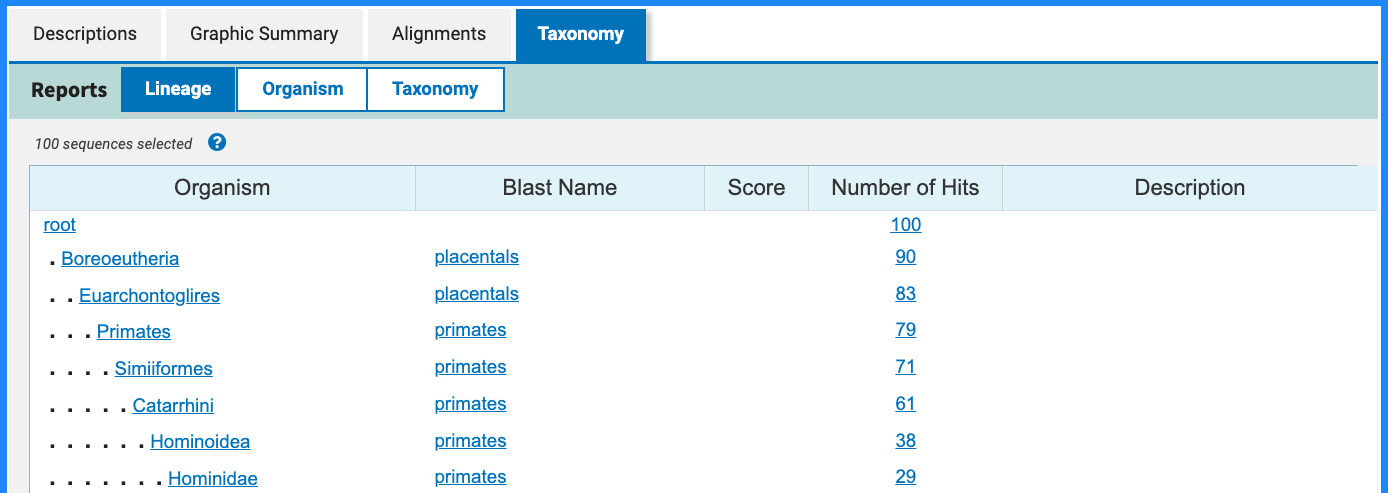
结果页的 “Taxonomy” 标签用于按物种谱系汇总 hits,便于快速判断匹配序列的物种分布与潜在污染来源。
5.4 如何下载与分享结果
在结果页面通常可以看到:
- Download 按钮:支持下载为 TSV、XML、FASTA 等多种格式,以便本地保存或进一步分析
- Send to:可将结果发送到文件、本地计算机、或保存到 NCBI 账号
- Request ID(RID):一个唯一的任务 ID,可以在一定时间内用来重新访问结果
六、常见问题与实战小技巧
6.1 常见报错与处理
- 提交后很久没有结果:
- 可能是数据库选择过大(如 nr)且序列较长,耐心等待
- 可尝试限制 Organism 或选择更小的数据库加快速度
- 提示序列格式错误:
- 检查是否包含非法字符(如中文、空格、特殊符号)
- 确保 FASTA ID 行以
>开头,后面不要再有>或 HTML 符号
- 结果为空或只有很差的匹配:
- 确认序列方向、是否含有大量 N(不确定碱基)
- 适当提高 E-value 阈值或减小 word size
6.2 如何根据研究目的调整参数
不同研究问题对 BLAST 的参数需求不同,可参考下表思路:
- Program:Highly similar sequences (megablast)
- E-value:默认或更严格,如 1e-10
- Database:nr/nt 或针对目标物种数据库
- Program:More dissimilar sequences 或使用 BLASTX/PSI-BLAST
- 适当提高 E-value 阈值以捕获更多远缘 hits
- 适当减小 word size 提高敏感性
- 推荐以蛋白序列为 Query,使用 BLASTP
- 多关注 Descriptions 中的功能注释和保守结构域
- 可结合 CDD(Conserved Domain Database)进行结构域扫描
完成本节练习后,可:
- 熟练打开 NCBI 在线 BLAST 平台,并选择合适的 BLAST 类型
- 用 FASTA 格式准备并提交核酸/蛋白序列
- 理解并调整常用参数(数据库、Organism、E-value、Program 等)
- 读懂 BLAST 结果中的图形概览、匹配列表和比对详情
建议在实际工作中多尝试不同的序列与参数组合,逐渐形成适合研究方向的 BLAST 使用习惯。
© 南京中医药大学 人工智能与信息技术学院 袁少勋