Seurat 是 R 语言中用于单细胞 RNA 测序(scRNA-seq)数据分析的主流工具包,由 Satija Lab 开发并维护。它覆盖从原始表达矩阵到细胞分群、细胞类型注释、差异表达与可视化的完整流程。
所有数据与中间结果都存放在一个 Seurat 对象 中。对象内包含:
RNA assay。其中 counts 为原始 count,data 为归一化后的表达,scale.data 为标准化后用于降维的高变基因矩阵。nCount_RNA(总 UMI)、nFeature_RNA(检测到的基因数)、percent.mt(线粒体基因占比)、seurat_clusters(聚类结果)等。pca、umap、tsne,用于后续绘图与聚类。Seurat 官方教程常用 PBMC 3K 作为示例数据集,便于在本地快速复现完整流程。
filtered_gene_bc_matrices,用 Read10X() + CreateSeuratObject() 创建对象;SeuratData 包安装 pbmc3k 后直接加载(InstallData("pbmc3k"),data("pbmc3k"))。经过聚类与注释后,PBMC 3K 中通常可区分出多种免疫细胞类型,例如:
教程会演示如何从原始矩阵得到这些细胞类型标签及对应的 Marker 基因。
SeuratData 的 pbmc3k 或教程提供的 R 代码生成“模拟”对象,重点学习对象结构与绘图函数。
使用 Seurat 做单细胞分析时,典型步骤包括:
CreateSeuratObject(),并(可选)计算 percent.mt。nFeature_RNA、nCount_RNA、percent.mt 过滤低质量细胞。NormalizeData()(默认 LogNormalize)。FindVariableFeatures()。ScaleData()(用于 PCA 的基因)。RunPCA() → RunUMAP() / RunTSNE()。FindNeighbors() + FindClusters()。下面按“质控 → 降维/分群 → 基因表达 → Marker/差异”顺序,说明 Seurat 中常用图的含义与对应函数。
| 图形 | 函数(示例) | 含义 |
|---|---|---|
| 小提琴图(QC 指标) | VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt")) |
展示每个细胞(或按分组)的基因数、总 UMI 数、线粒体比例分布,用于设定质控阈值、发现异常样本。 |
| 散点图(基因数 vs UMI) | FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA") |
看 nCount 与 nFeature 的相关性,通常呈正相关;离群点可能是双细胞或破损细胞。 |
| 图形 | 函数(示例) | 含义 |
|---|---|---|
| UMAP / t-SNE 分群图 | DimPlot(pbmc, reduction = "umap", group.by = "seurat_clusters") |
在 UMAP(或 t-SNE)二维空间中按聚类或注释标签着色,展示细胞群分布与分离程度。 |
| PCA 双标图 | DimPlot(pbmc, reduction = "pca") 或 VizDimLoadings() |
展示主成分上的细胞分布或各基因对主成分的贡献(loading),用于理解驱动差异的主成分。 |
| 肘部图 | ElbowPlot(pbmc) |
各主成分解释的方差比例,用于选择保留的 PC 数量(通常取“肘部”附近)。 |
| 图形 | 函数(示例) | 含义 |
|---|---|---|
| 降维图上的基因表达 | FeaturePlot(pbmc, features = "CD3D") |
在 UMAP/t-SNE 上用颜色表示某个基因的表达量,用于看该基因在哪些细胞/区域高表达,辅助细胞类型判断。 |
| 小提琴图(基因表达) | VlnPlot(pbmc, features = "CD3D", group.by = "seurat_clusters") |
按 cluster(或细胞类型)展示某基因表达量分布,比较不同群之间表达差异。 |
| 脊线图 | RidgePlot(pbmc, features = "CD3D", group.by = "seurat_clusters") |
类似小提琴图,以脊线形式展示各群中某基因的表达分布,适合多群对比。 |
| 图形 | 函数(示例) | 含义 |
|---|---|---|
| 点图(DotPlot) | DotPlot(pbmc, features = marker_genes) + RotatedAxis() |
横轴为基因,纵轴为 cluster(或细胞类型);点大小表示表达该基因的细胞比例,颜色表示平均表达量,用于快速对比多个 Marker 在各群中的表达。 |
| 热图(DoHeatmap) | DoHeatmap(pbmc, features = top_markers, size = 3) |
对每个 cluster 的 top marker 基因做热图,行是基因、列是细胞(常按 cluster 排列),颜色为标准化表达,用于展示各群特征基因表达模式。 |
| 气泡图(比例 + 表达) | 同上 DotPlot,有时与 scale = TRUE 等参数联用 |
与 DotPlot 一致,强调“表达比例”与“平均表达”两个维度,是 Marker 展示最常用的图之一。 |
上述图中,DimPlot、FeaturePlot、VlnPlot、DotPlot、DoHeatmap 是 Seurat 教程中最常出现的五类图,分别对应:分群展示、基因在空间上的表达、基因在各群分布、多基因多群对比、Marker 热图。
每一小节为「一段说明 + 一段代码」,按顺序复制到 R 中运行即可。建议先在 RStudio 中新建项目(见 6.0),再完成 6.1 环境与数据,最后按 6.2~6.5 做质控、流程、出图。
新建项目便于统一管理脚本、数据和结果,且每次打开项目即进入对应工作目录。项目名称建议与本次分析相关,例如 seurat-pbmc(Seurat + PBMC 示例)或 single-cell-seurat。
seurat-pbmc(或 single-cell-seurat);Create project as subdirectory of 选择你想保存的父文件夹,点击 Create Project。创建完成后,RStudio 会打开该项目,工作目录即为该文件夹。后续可将 R 脚本保存于此,运行时代码中的相对路径均相对于该目录。
先设置 CRAN 为国内镜像(如清华),再安装 Seurat,可显著加快下载。只需在首次使用或更新包时执行。
options(repos = c(CRAN = "https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
install.packages("Seurat")
SeuratData 提供官方示例数据。先从 GitHub 安装 SeuratData 包,再下载 pbmc3k 数据集(约 2,700 细胞)。若 GitHub 较慢,可多试几次或配置网络。
if (!requireNamespace("remotes", quietly = TRUE)) install.packages("remotes")
remotes::install_github("satijalab/seurat-data")
library(SeuratData)
InstallData("pbmc3k")
每次打开 R 做单细胞分析前,先加载 Seurat 与 SeuratData。
library(Seurat) library(SeuratData)
将内置的 pbmc3k 数据载入内存,并统一用 pbmc 作为对象名。若出现 “Please run UpdateSeuratObject on your object”,说明对象是旧版 Seurat 格式,需运行 UpdateSeuratObject(pbmc) 转为当前版本后再继续。
data("pbmc3k")
pbmc <- pbmc3k
pbmc <- UpdateSeuratObject(pbmc)
pbmc3k 的 meta 里默认没有 percent.mt,需要先算出来,后面的质控小提琴图才能画三张(含线粒体比例)。线粒体基因名通常以 MT- 开头。
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
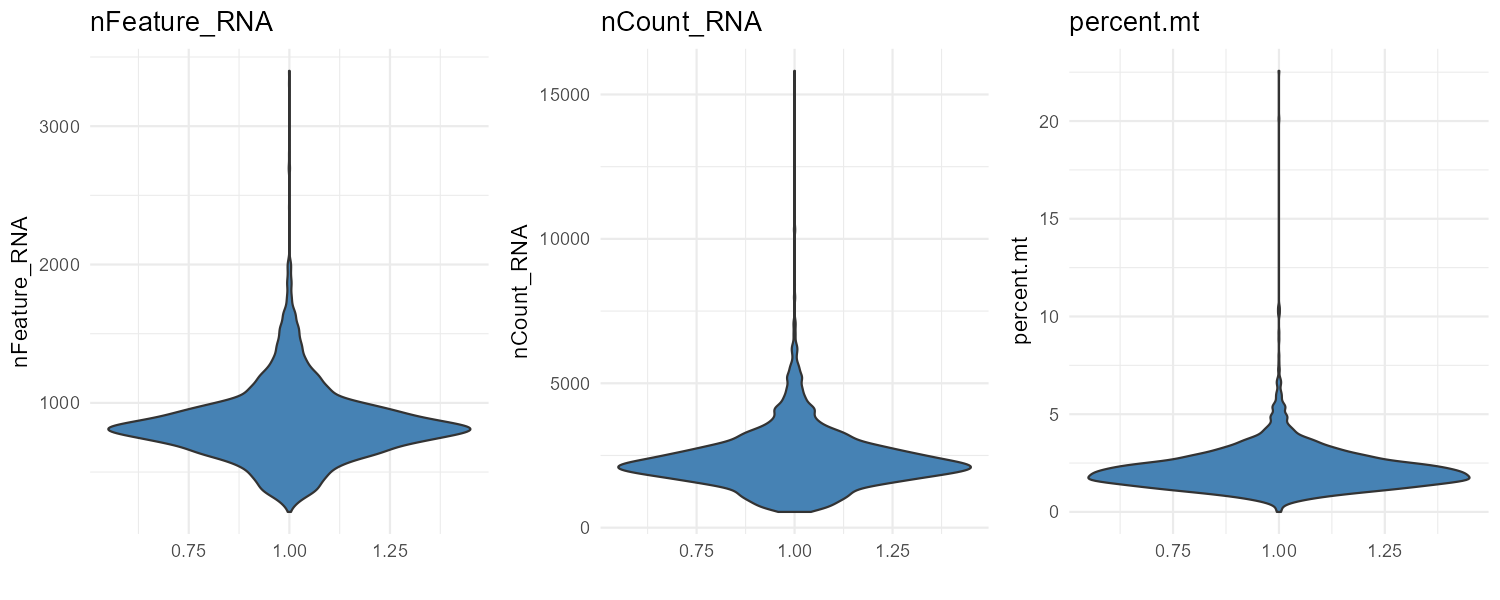
查看每个细胞检测到的基因数、总 UMI 数、线粒体基因占比的分布,用于设定过滤阈值或发现异常样本。若只有两幅图、提示 variables were not found: percent.mt,请先运行上面 6.1.5 计算 percent.mt。
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
结果解读:可据此观察基因数、UMI 数、线粒体比例的分布范围与离群点,从而设定质控阈值(如 nFeature_RNA 200–2500、percent.mt < 5%)并过滤低质量细胞。
看总 UMI 与检测基因数的相关性;离群点可能是双细胞或破损细胞。
FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
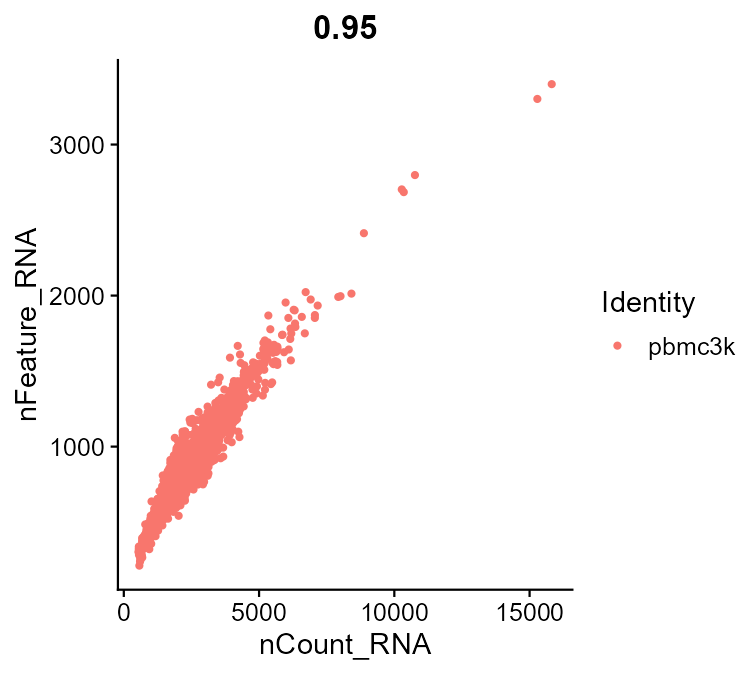
结果解读:nCount 与 nFeature 通常呈正相关;若出现明显偏离主云团的点,可能是双细胞或破损细胞,应在质控时剔除。
对 raw count 做对数归一化,使细胞间可比。默认方法为 LogNormalize。
pbmc <- NormalizeData(pbmc)
筛选在细胞间变异较大的基因,用于后续降维与聚类,默认取 2000 个。
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
对高变基因做中心化与缩放,得到 scale.data,供 PCA 使用。
pbmc <- ScaleData(pbmc)
在高变基因的 scale 矩阵上做 PCA,得到低维表示。后续用前若干主成分做聚类与 UMAP。
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
基于前 10 个主成分构建 KNN 图,再用 Louvain 等算法得到细胞聚类标签(seurat_clusters)。resolution 越大簇越多。
pbmc <- FindNeighbors(pbmc, dims = 1:10) pbmc <- FindClusters(pbmc, resolution = 0.5)
在 PCA 基础上计算 UMAP 二维坐标,用于可视化分群与基因表达。
pbmc <- RunUMAP(pbmc, dims = 1:10)
在 UMAP 空间中展示各聚类,每点一个细胞,颜色为 seurat_clusters。
DimPlot(pbmc, reduction = "umap", group.by = "seurat_clusters")
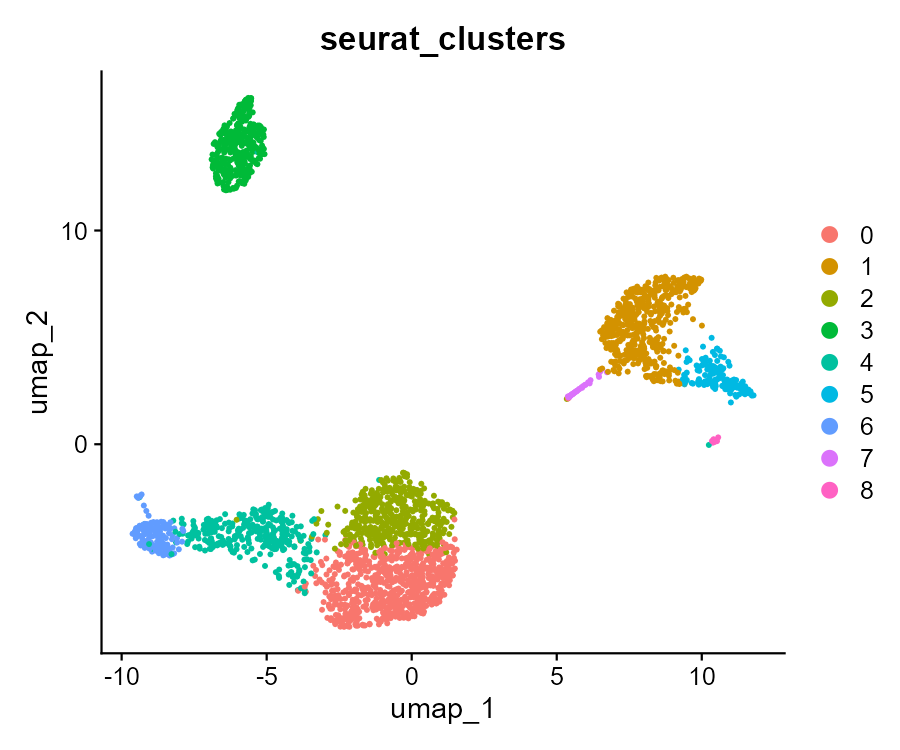
结果解读:不同颜色代表不同 cluster,可直观看出细胞在 UMAP 空间中的分群是否清晰、是否有过度分裂或合并,为后续细胞类型注释提供空间依据。
在 UMAP 上用颜色表示指定基因的表达量,用于判断该基因在哪些细胞/区域高表达。
FeaturePlot(pbmc, features = c("CD3D", "CD14", "MS4A1"))
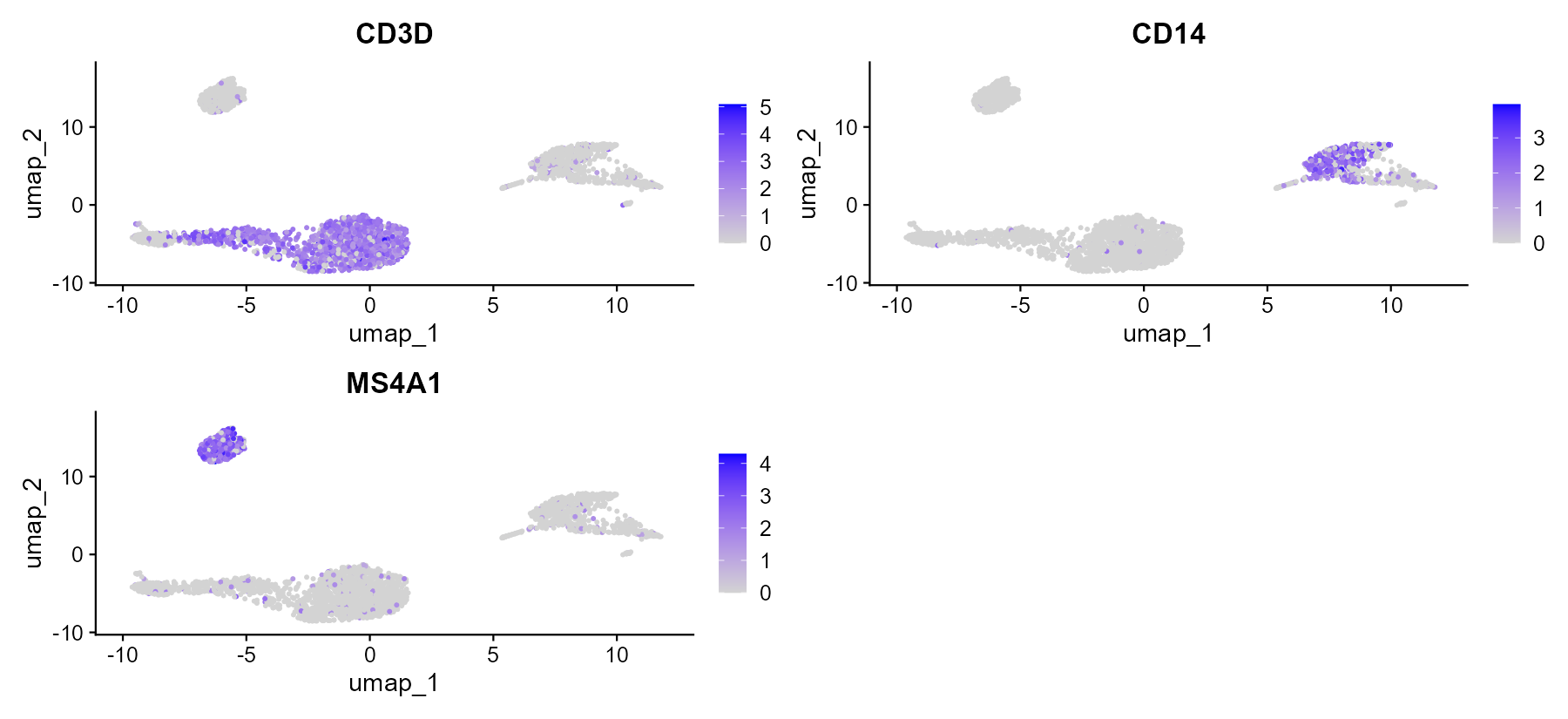
结果解读:CD3D 高表达区对应 T 细胞群,CD14 高表达区对应单核细胞群,MS4A1 高表达区对应 B 细胞群,可与分群图结合判断各 cluster 的细胞类型。
按聚类展示某基因的表达分布,比较不同群之间的表达差异。
VlnPlot(pbmc, features = "CD3D", group.by = "seurat_clusters")
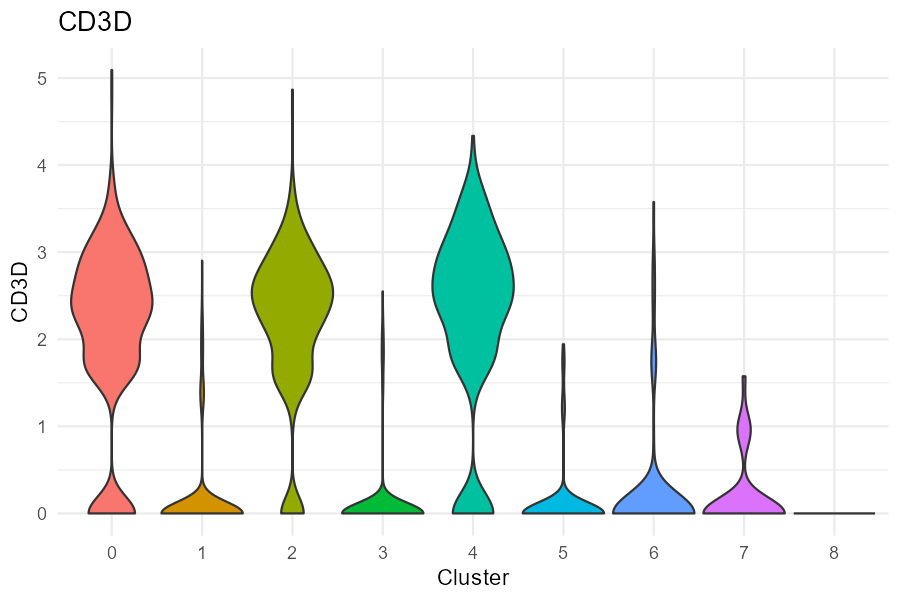
结果解读:CD3D 在部分 cluster 中明显高表达、在其余 cluster 中较低,可帮助确认哪些 cluster 为 T 细胞相关,并评估 Marker 的特异性。
与小提琴图类似,以脊线形式展示各群中某基因的表达分布。
RidgePlot(pbmc, features = "CD3D", group.by = "seurat_clusters")
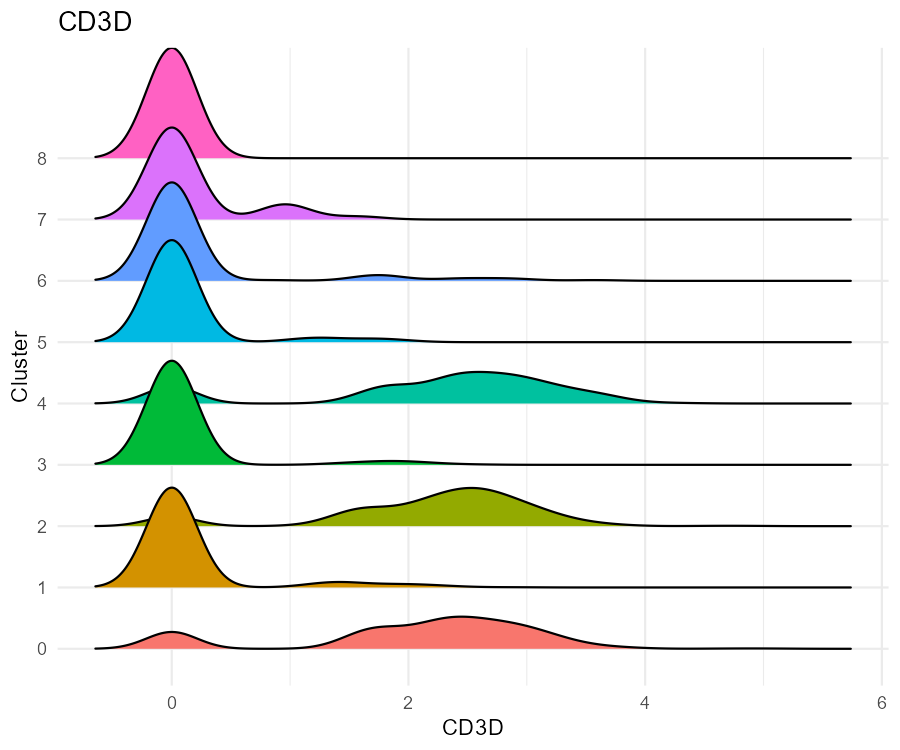
结果解读:脊线峰的位置与高度反映各 cluster 中 CD3D 表达水平,便于在多群并排时快速比较 T 细胞相关 cluster 与其他群的差异。
对每个聚类做差异表达,得到该群相对其他群高表达的基因(只保留正标记、满足最小表达比例与 logFC 阈值)。
markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
按 cluster 和 avg_log2FC 排序,每个 cluster 保留前 10 个基因,供 DotPlot/DoHeatmap 使用。
top10 <- markers[order(markers$cluster, -markers$avg_log2FC), ] top10 <- do.call(rbind, lapply(split(top10, top10$cluster), head, 10)) top_genes <- unique(top10$gene)
横轴为基因、纵轴为 cluster;点大小表示表达比例,颜色表示平均表达量,便于对比多基因在多群中的表达。
DotPlot(pbmc, features = top_genes) + RotatedAxis()
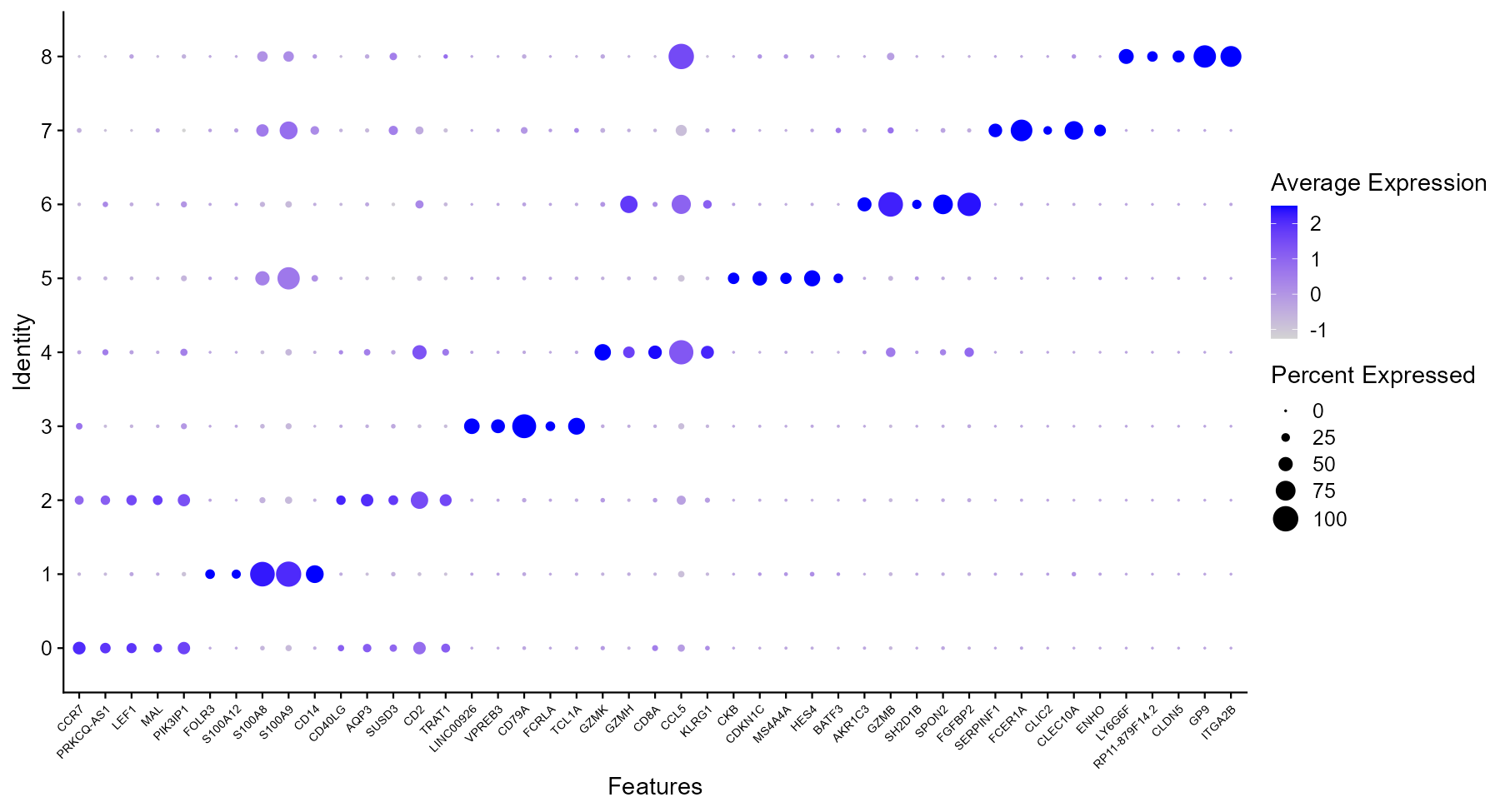
结果解读:点大且颜色深的基因在该 cluster 中表达比例高、平均表达强,可据此快速识别各群的特征 Marker,辅助细胞类型命名与验证。
对上述 top 基因做热图,行是基因、列是细胞(按 cluster 排列),展示各群特征表达模式。
DoHeatmap(pbmc, features = top_genes, size = 3)
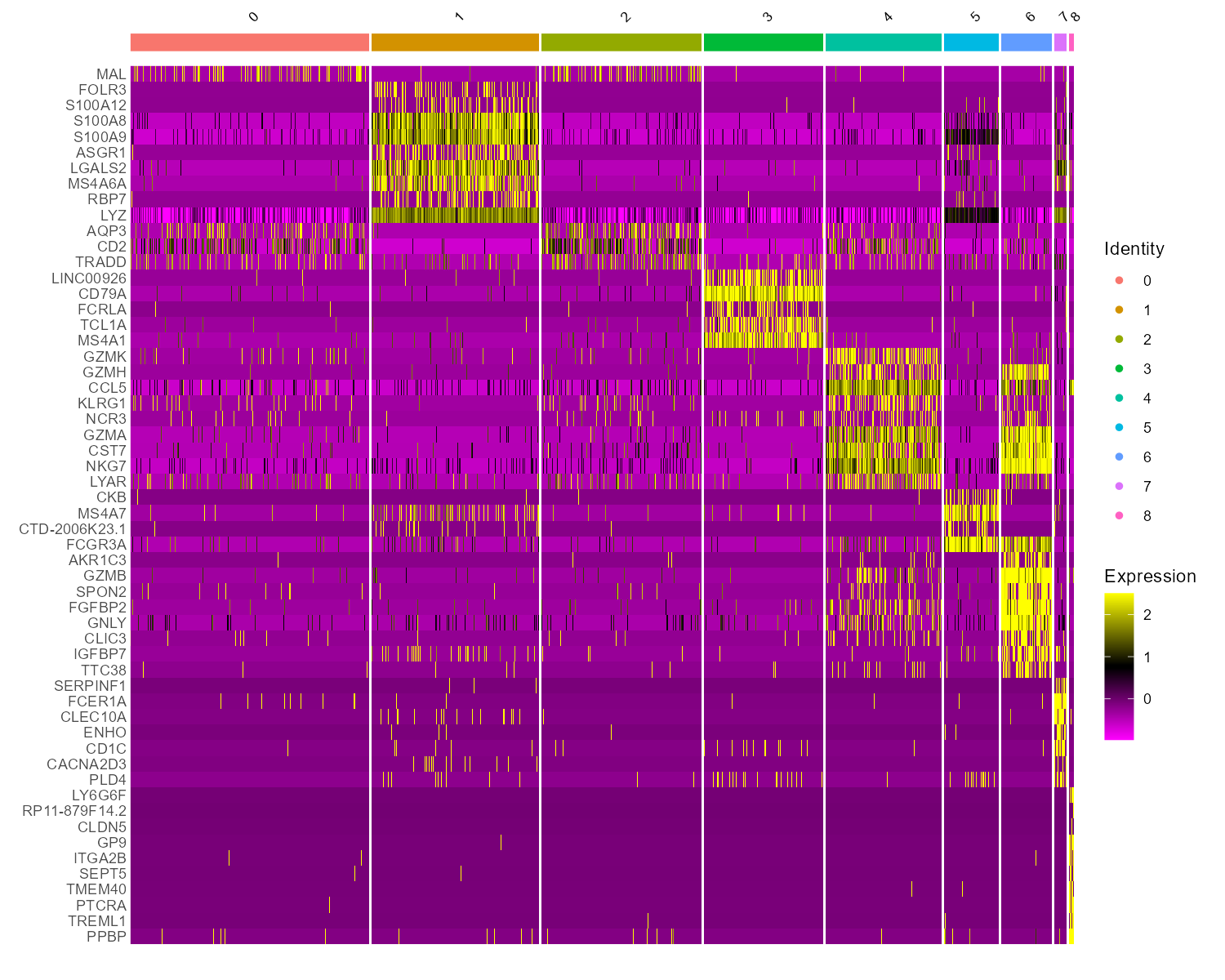
结果解读:每列为一个细胞、每行为一个 Marker 基因,颜色表示标准化表达;可看出各 cluster 对应的基因表达模式,用于确认分群与注释是否合理。
scripts/seurat_generate_figures.R 生成(需已安装 Seurat 与 SeuratData 并成功加载 pbmc3k)。在项目根目录执行 Rscript scripts/seurat_generate_figures.R 即可将图片写入 docs/images/seurat/。若 pbmc3k 已通过 InstallData("pbmc3k") 安装,只需运行 6.1.3、6.1.4 即可加载。调整 FindClusters(..., resolution = 0.5) 中的 resolution(如 0.3~1.0)可改变聚类粗细;可根据已知 Marker 将 seurat_clusters 重命名为细胞类型后再绘图。