分子进化树的构建
一、MEGA 的介绍与官方网站
1.1 软件简介
MEGA(Molecular Evolutionary Genetics Analysis)是常用的分子进化与系统发育分析软件,提供序列比对、进化模型选择、邻接法与最大似然法等多种建树途径,以及树形编辑与统计检验等图形化功能。实验桌面端以 Windows 安装包最为常见;其他系统可在官网选择对应版本。
下载与安装前请准备可联网的浏览器;安装过程可能需要本机管理员权限(视系统策略而定)。使用软件须遵守官网公示的许可条款,并限于教学与科研等允许用途。
1.2 官方网站
MEGA 由开发团队通过官方网站发布版本与文档,请认准下列入口,避免从不明第三方站点获取安装包。
二、MEGA 的下载与安装
以下以在 Windows 上从官网获取 .exe 安装包并完成安装为例;若官网页面选项与下文略有出入,以当前版本页面为准。
视频教程:MEGA 下载与安装(MP4)
2.1 打开主页并进入下载流程
- 在浏览器中打开 MEGA 官方主页:https://www.megasoftware.net/。
- 在主页上找到绿色的 DOWNLOAD 按钮,单击进入下载相关页面。
2.2 接受用户许可协议
- 页面将跳转至 MEGA User Agreement(用户协议)。
- 阅读后在页面下方单击绿色的 Accept(同意),继续下载流程。
2.3 填写基本信息并下载安装包
- 在正式下载前,按页面要求填写必要信息:例如在 Country(国家/地区)中选择所在地(如 China),并填写机构名称与身份等字段(以页面实际选项为准)。
- 填写完毕后,单击页面上的下载按钮,将适用于本机的安装包保存到磁盘(Windows 一般为
.exe)。
若国际网络访问较慢,可在遵守许可的前提下使用课程提供的离线安装包或单位镜像。
2.4 运行安装程序与安装向导(Windows)
- 在资源管理器中双击已下载的
.exe。若出现用户账户控制(UAC)提示,在确认文件来源可靠后选择允许。 - 语言:向导首屏通常可选择界面语言(默认多为 English),单击 OK。
- 许可条款:勾选同意安装协议,单击 Next。
- 安装路径:可使用默认路径,或通过 Browse 指定目录,然后继续 Next。
- 其后按向导提示依次单击 Next,直至开始复制文件并完成安装阶段(具体页数因版本可能略有不同)。
2.5 完成安装并启动
- 待进度结束,在结束页单击 Finish。若向导提供立即运行选项,可按需勾选。
- 也可从开始菜单或桌面快捷方式启动 MEGA。
- 确认 图形化主界面正常打开后,即可进行后续的序列导入、比对与系统发育树构建等操作。
以上为从 MEGA 官方网站获取安装包、完成注册与下载,以及在 Windows 环境下安装并启动主界面的完整流程。
三、示例数据:INS 基因 CDS
3.1 数据文件与说明
本教程后续将以 多个哺乳动物(含部分非哺乳类对照)胰岛素基因(INS)CDS 的 FASTA 文件为例,演示在 MEGA 12 中完成:导入序列 → 多序列比对 → 构建系统发育树 → 导出结果。
- 数据文件:
data/INS_cds.fasta - 同一物种可能包含多个转录本(如
human_tx1、human_tx2等)。
3.2 下载示例文件
- 下载:INS_cds.fasta
- 仓库路径:
data/INS_cds.fasta
四、MEGA构建分子进化树
本节以第 3 章的 INS CDS 为例,给出 MEGA 12 的常用操作路径。不同版本的菜单名称可能略有差异,但整体流程一致:导入 → 比对 → 选择方法 → 建树 → 导出。
视频教程:MEGA 12 构建分子进化树(MP4)
4.1 导入序列
- 启动 MEGA 12。
- 在主界面选择 Align(序列比对相关入口),进入比对工作区(Alignment Explorer)。
- 在比对窗口中选择 Edit 或 Data 菜单下的导入功能(不同界面位置可能不同),从文件导入
data/INS_cds.fasta。 - 确认每条序列的名称(如
human_tx1、mouse_tx1)在左侧列表可见,且序列方向一致(均为 5′→3′)。
4.2 多序列比对
- 在 Alignment Explorer 中选择 Align by MUSCLE(或课程指定的 ClustalW/Clustal Omega),对所有序列进行多序列比对。
- 比对完成后,检查是否出现明显异常:例如某条序列前后出现大量缺口(gap),或长度显著不同导致比对质量下降。
- 将比对结果保存为 MEGA 格式工程文件(常用为
.meg),以便后续反复调整参数。
4.3 修剪与质量检查
- 若序列两端存在明显的低一致性区域或长度不齐,可在比对窗口中对两端进行适度修剪,使大多数序列覆盖到相同的比对区段。
- 确认最终用于建树的数据为“已比对”的序列集合,而不是原始未比对 FASTA。
4.4 选择建树方法与参数
- 在 MEGA 主界面选择 Phylogeny(系统发育)相关入口,基于已对齐的数据进行建树。
- 常用方法:
- Neighbor-Joining(NJ):速度快,适合课堂演示整体流程。
- Maximum Likelihood(ML):更常用于正式分析,计算时间更长。
- 模型与缺口处理:对于核酸 CDS,通常选择核酸替换模型(以课程要求为准);缺口(gap)与缺失位点常见处理为 Complete Deletion 或 Partial Deletion(设置阈值保留尽可能多的位点)。
- 置信度评估:勾选 Bootstrap(自助法)并设置重复次数(课堂演示可 100;更稳定的分析常用 500–1000)。
- 运行建树,得到系统发育树窗口(Tree Explorer)。
4.5 导出与展示
- 在 Tree Explorer 中调整树形显示:分支长度、节点置信度(bootstrap 值)显示位置、字体与标签等。
- 导出树文件与图片:常见格式包括 Newick(
.nwk)用于后续软件复用,以及 PNG/SVG 用于报告与课件。 - 建议保存当前工程(比对文件 + 树文件),以便下一次修改参数后对比不同方法(NJ vs ML)、不同缺口处理策略的影响。
五、使用 R 语言构建进化树
除 MEGA 外,也可用 R 读取同一套 INS CDS FASTA,完成比对、距离计算、建树与可视化。下面给出配套脚本下载、流程概览与示例输出图。
5.1 R 脚本下载
- 下载:phylogenetic_tree.R
- 仓库路径:
data/phylogenetic_tree.R
脚本默认与 INS_cds.fasta 位于同一目录时可直接运行;若路径不同,请修改脚本中的 input_fasta。
5.2 流程概览(顺序流程图)
下图自上而下为脚本执行顺序;箭头表示步骤先后关系。
- 安装并加载 R 包:
Biostrings、muscle、ape、ggtree、ggplot2、grid等;首次使用需在 R 中按各包说明完成安装。 - 读取序列:用
readDNAStringSet()读入INS_cds.fasta。 - 多序列比对:
muscle::muscle()比对后转为DNAbin,供距离计算使用。 - 距离与建树:
ape::dist.dna()采用 TN93 模型、pairwise.deletion = TRUE;再用ape::fastme.bal()得到带分支长度的树。 - 定根:用共表型距离找出最远的一对 tip,取其一作为外群,
ape::root(..., resolve.root = TRUE)。 - 标签与分组着色:将 tip 名映射为拉丁学名与中文注释,并按灵长类、啮齿类等分类群赋色。
- 出图与保存:
ggtree绘制分支长度标尺与图例,ggsave()输出高分辨率INS_tree.png。
5.3 步骤说明与结果图
按上表顺序在 R 或 RStudio 中运行脚本后,应得到与下图类似的系统发育树(横轴为遗传距离,叶节点按分类群着色)。
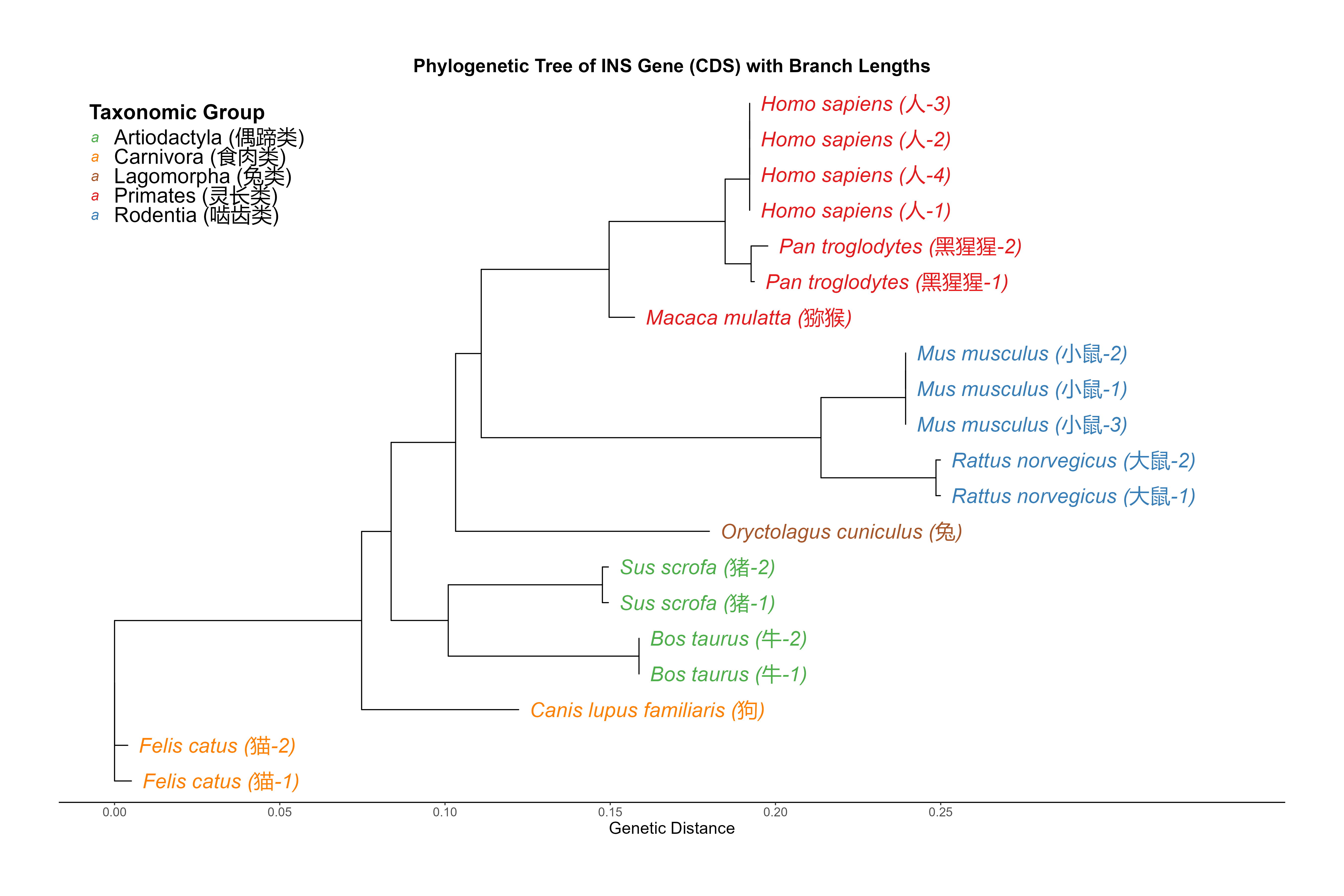
INS_cds.fasta 的 INS 基因 CDS 系统发育树(R 脚本输出示例)。六、作业与拓展
- 必做:使用本页示例数据(或课堂指定数据)在 MEGA 12 中完成一次建树,并提交:
① 一张导出的树图(PNG/SVG 均可);
② 用 1–2 句话写出你从树上观察到的一个现象(例如:哪些物种聚成一支、某个节点支持率高/低等)。 - 选做:在同一套已对齐序列上,分别使用 Maximum Likelihood(最大似然法)、Neighbor-Joining(邻接法)、Minimum Evolution(最小进化法) 构建三棵进化树,并比较:三棵树的主要分支拓扑是否一致?哪些节点的支持率(如 bootstrap)差异最明显?
- 拓展:导出 Newick(
.nwk)并导入任意树可视化软件(如 iTOL、FigTree 等),尝试对某一类群着色并保存一张排版后的成图。