02 · 数据质量评估与数据清洗
一、医学数据基础
医学数据是进行数据分析与挖掘的起点。理解医学数据的类型、特征与来源,有助于制定合理的数据质量策略与分析方法。
1.1 医学数据类型与特点
医学数据可以按结构化程度、大致分为结构化数据与非结构化数据,两类数据在采集方式、存储形式和分析方法上差异较大。
-
结构化数据
以“行 × 列”的表格形式存在,字段含义清晰、单位明确,便于直接做统计与建模。
常见示例:
- 住院电子病历首页:
patient_id、admission_date、discharge_date、primary_diagnosis_code等字段 - 检验指标表:每行一条检验结果,包含
test_name(如血红蛋白)、result_value、unit、reference_range、sample_time - 用药记录表:
drug_name、dose、route(口服/静滴)、frequency(bid/q8h)、start_time、end_time
- 住院电子病历首页:
-
非结构化数据
缺乏固定字段,内容多以自由文本或图像/序列形式存在,需要额外的文本挖掘、影像分析或序列分析方法。
常见示例:
- 病历文本:门诊病历、出院小结、手术记录中的自然语言描述,如“患者主诉胸闷 3 天,加重 1 天”
- 医学影像:CT/MRI/超声图像以及对应的报告文本,需要通过影像分割、检测等方法提取特征
- 组学与基因序列:如
FASTQ/BAM文件中的序列数据,或转录组/蛋白组表达矩阵
-
医学数据 5V 特征
医学大数据常沿用通用大数据的“5V”框架来概括其规模与性质,在质量评估与系统设计时需同时考虑这五方面。
- 海量(Volume)单家三甲医院每年即可产生 TB 级诊疗与影像数据;区域平台、国家队列或组学库往往达到 PB 级,对存储与计算提出高要求。
- 多样(Variety)既有结构化表格(病案首页、检验结果),也有病历文本、影像、波形、基因序列等多模态数据,需要不同的采集、存储与分析手段。
- 高价值(Value)数据直接关联诊疗决策、科研发现与公共卫生政策,价值密度高,但也涉及隐私与伦理,需在利用与保护之间取得平衡。
- 实时(Velocity)重症监护、手术监护、急诊等场景下数据持续产生且需近实时告警;流行病监测、不良事件上报等也要求及时汇聚与响应。
- 准确性(Veracity)医学结论依赖数据真实性与一致性;录入错误、缺失、编码不统一等会直接影响诊断、统计与模型效果,因此数据质量与可信度至关重要。
例如:区域医疗大数据平台中,来自多家医院的门急诊记录、检查检验、影像和随访信息持续接入,形成 TB 级乃至更大规模的时序数据,同时具备多模态(表格+文本+影像)、高价值(支撑临床与科研)和实时性(部分需近实时分析)等特点,并在整合过程中面临准确性、一致性等质量挑战。
-
医学数据特殊性
冗余、多属性、不完整、强时序性。
例如:同一患者在不同医院、不同信息系统中有多份病历(冗余)、一次住院包含数百个实验室指标与用药记录(多属性)、长期随访中存在多次失访(不完整)、生命体征呈时间序列随病程变化(强时序性)。
1.2 医学数据来源
医学数据来源非常广泛,既包括医院内部产生的诊疗数据,也包括科研、公共卫生以及可穿戴设备等新兴渠道。
- 电子病历(EHR/EMR):入院记录、病程记录、体格检查、出院小结等结构化 + 文本信息
- 检验/检查系统(LIS/RIS/PACS):血常规、生化、心电图、CT/MRI 报告及影像文件
- 护理记录系统:生命体征曲线、输液记录、压疮风险评估、疼痛评分等
- 随机对照临床试验(RCT)数据:严格的入排标准、预先定义的结局指标和随访计划
- 队列研究数据:随时间跟踪人群的暴露、结局和混杂因素,如心血管队列、肿瘤生存队列
- 生物样本库:与血液、组织样本对应的临床信息和组学检测结果
- 公共卫生监测系统:传染病疫情报告、慢病登记(肿瘤登记、卒中登记)
- 医保结算数据:门急诊费用、用药和诊疗项目编码,可用于疾病负担和卫生经济学分析
- 开放医学数据库:如 MIMIC、PhysioNet 等 ICU 生理监测和病历数据集
- 可穿戴设备:心率、步数、睡眠时长、心电贴监测的房颤事件
- 移动医疗 App:血糖自测上报、用药提醒完成情况、远程随访问卷
- 家庭监测设备:家用血压计、血糖仪上传的长期家庭自测数据
二、医学数据质量分析
数据质量分析是数据预处理的前提,主要任务是识别"脏数据"及其成因,并系统梳理数据质量问题的类型与识别方法。
2.1 脏数据及其原因和案例
"脏数据"指不符合数据质量要求的数据,会直接影响统计结果和模型性能。常见成因与案例如下:
下面以 R 语言内置数据集 Melanoma(MASS 包)为例:先加载并查看该数据集,再在其上人为构造上述四类脏数据,便于后续识别与清洗。
完整操作代码(国内环境适配版)
# ========== 第一步:安装 MASS 包 ==========
# 临时设置国内镜像(清华镜像,解决下载慢/失败问题)
options(repos = c(CRAN = "https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
install.packages("MASS") # 如果已安装,会提示无需安装,不影响运行
# ========== 第二步:加载 MASS 包 ==========
library(MASS) # 每次重启 R 都需要执行
# ========== 第三步:加载并查看 Melanoma 数据集 ==========
# 方式1:显式加载(推荐新手)
data(Melanoma)
cat("=== Melanoma 数据集前 6 行 ===\n")
head(Melanoma)
cat("\n=== Melanoma 数据集结构(变量类型与含义) ===\n")
str(Melanoma)
cat("\n=== Melanoma 数据集基本统计信息 ===\n")
summary(Melanoma)
# 方式2:加载包后也可直接使用 Melanoma,无需再调用 data()在 Melanoma 上构造四类脏数据
复制副本
Melanoma_dirty <- as.data.frame(Melanoma)缺失数据:随机将部分 age、thickness 置为 NA
set.seed(123)
n <- nrow(Melanoma_dirty)
Melanoma_dirty$age[sample(n, 8)] <- NA
Melanoma_dirty$thickness[sample(n, 6)] <- NA异常数据:人为写入超出合理范围的值
Melanoma_dirty$age[3] <- -1
Melanoma_dirty$age[10] <- 200
Melanoma_dirty$thickness[5] <- 999重复数据:追加若干行完全重复的记录
dup_rows <- Melanoma_dirty[c(1, 1, 15, 15, 15), ]
Melanoma_dirty <- rbind(Melanoma_dirty, dup_rows)不一致数据:sex 混用 0/1 与 "男"/"女"
Melanoma_dirty$sex <- as.character(Melanoma_dirty$sex)
Melanoma_dirty$sex[Melanoma_dirty$sex == "0"] <- "女"
Melanoma_dirty$sex[Melanoma_dirty$sex == "1"] <- "男"
Melanoma_dirty$sex[sample(which(Melanoma_dirty$sex == "男"), 3)] <- "1"
Melanoma_dirty$sex[sample(which(Melanoma_dirty$sex == "女"), 2)] <- "0"验证脏数据版
head(Melanoma_dirty, 10)
cat("原始行数:", nrow(Melanoma_dirty), " 去重后:", nrow(unique(Melanoma_dirty)), "\n")
cat("age NA 数:", sum(is.na(Melanoma_dirty$age)), " thickness NA 数:", sum(is.na(Melanoma_dirty$thickness)), "\n")具体案例示例:
- 在心衰住院患者数据库中,约有 15% 的病例缺少
BNP指标,其中多数为病情较轻的患者(选择性未检),属于典型的 MAR/MNAR 缺失。 - 某院门诊系统中,部分身高录成“1700 cm”或“1.7 cm”,与正常身高范围严重不符,属于单位/录入错误导致的异常数据。
- 同一患者在转科过程中,多次复制住院记录,导致相同住院号下存在 3 条完全重复的入院信息,属于重复数据。
- 在多中心研究中,一家医院使用 ICD-10 编码“E11.900”表示 2 型糖尿病,另一家只用汉字“2 型糖尿病”,如果不做统一映射,就会出现不一致数据。
2.2 数据质量问题识别
识别数据质量问题,通常结合统计方法与业务规则两条线并行开展。
- 缺失值通过
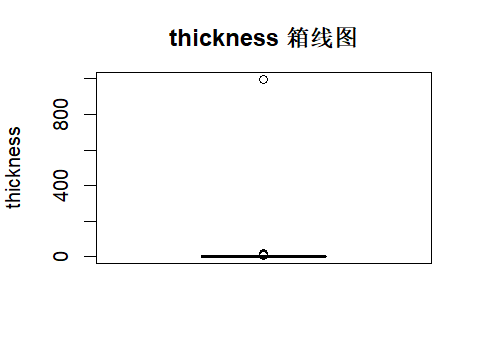
is.na()、缺失比例统计和缺失模式可视化区分 MCAR / MAR / MNAR。 - 噪声与异常值利用分布统计(均值、标准差、分位数)、箱线图、散点图以及 3σ/IQR 等规则识别可疑点。
- 重复数据基于业务主键(如患者 ID + 就诊号 + 检查时间)统计重复记录数,区分完全重复与部分重复。
- 不一致性通过枚举值频次、单位检查和逻辑校验(如出院时间 >= 入院时间)识别异常。
以下均基于前文构造的 Melanoma_dirty,对四类问题分别给出识别代码。
(1)缺失值识别
各列缺失个数与缺失比例
na_count <- colSums(is.na(Melanoma_dirty))
na_pct <- na_count / nrow(Melanoma_dirty) * 100
data.frame(缺失个数 = na_count, 缺失比例 = round(na_pct, 2))
na_count[na_count > 0]缺失模式(age / thickness 交叉表,便于判断 MCAR/MAR/MNAR)
table(age_缺失 = is.na(Melanoma_dirty$age), thickness_缺失 = is.na(Melanoma_dirty$thickness))(2)噪声与异常值识别
连续变量分布:快速发现异常
summary(Melanoma_dirty$age)
summary(Melanoma_dirty$thickness)3σ 法 / IQR 法标记异常行
age_ok <- Melanoma_dirty$age[!is.na(Melanoma_dirty$age)]
mu <- mean(age_ok); s <- sd(age_ok)
which(Melanoma_dirty$age < (mu - 3*s) | Melanoma_dirty$age > (mu + 3*s))
th <- Melanoma_dirty$thickness
q1 <- quantile(th, 0.25, na.rm = TRUE); q3 <- quantile(th, 0.75, na.rm = TRUE)
iqr <- q3 - q1
which(th < (q1 - 1.5*iqr) | th > (q3 + 1.5*iqr))箱线图可视化
boxplot(Melanoma_dirty$age, main = "age 箱线图")
boxplot(Melanoma_dirty$thickness, main = "thickness 箱线图")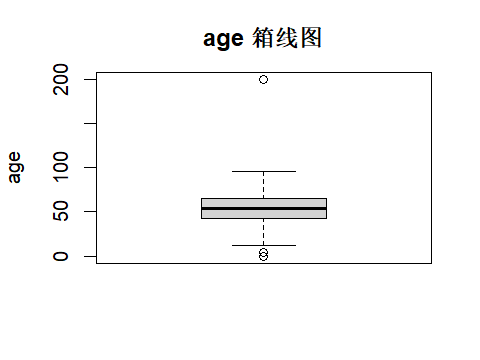
(3)重复数据识别
按整行完全重复统计
n_total <- nrow(Melanoma_dirty)
n_unique <- nrow(unique(Melanoma_dirty))
cat("总行数:", n_total, " 去重后:", n_unique, " 重复行数:", n_total - n_unique, "\n")按业务主键(time + age + thickness + sex)看重复
library(dplyr)
dup_key <- Melanoma_dirty %>%
count(time, age, thickness, sex) %>%
filter(n > 1)
dup_key(4)不一致性识别
枚举值频次:检查 sex、status 编码是否混用
table(Melanoma_dirty$sex, useNA = "ifany")
table(Melanoma_dirty$status, useNA = "ifany")逻辑校验:age 合理范围
bad_age <- Melanoma_dirty$age < 0 | Melanoma_dirty$age > 120
bad_age[is.na(bad_age)] <- FALSE
which(bad_age)三、医学数据清洗
数据清洗是数据预处理的核心步骤,包括对单表/单源数据的清洗与多源数据的集成、转换及特征工程。
3.1 数据清洗
围绕完整性、准确性、一致性与可分析性,通常开展以下四类处理:
- 缺失值处理根据缺失机制选择删除、简单填充、KNN 插值或模型预测等方法。
- 异常值处理结合统计规则(3σ、IQR)、可视化和专业知识剔除或修正异常。
- 重复数据处理定义合理主键,进行去重、记录合并及冲突解决。
- 数据标准化统一单位与编码,使多源数据可比、可整合。
(1)缺失值处理
方式 A:直接删除含缺失的行
clean_missing <- na.omit(Melanoma_dirty)方式 B:中位数填充(保留样本量)
clean_missing <- as.data.frame(Melanoma_dirty)
clean_missing$age[is.na(clean_missing$age)] <- median(clean_missing$age, na.rm = TRUE)
clean_missing$thickness[is.na(clean_missing$thickness)] <- median(clean_missing$thickness, na.rm = TRUE)验证缺失个数
cat("age NA 数:", sum(is.na(clean_missing$age)), " thickness NA 数:", sum(is.na(clean_missing$thickness)), "\n")(2)异常值处理
领域规则:age [0,120]、thickness 合理上限,超出置 NA
d <- as.data.frame(clean_missing)
d$age[d$age < 0 | d$age > 120] <- NA
d$thickness[d$thickness > 50] <- NAIQR 法:箱外置 NA
th <- d$thickness[!is.na(d$thickness)]
q1 <- quantile(th, 0.25); q3 <- quantile(th, 0.75); iqr <- q3 - q1
out_iqr <- (d$thickness < (q1 - 1.5*iqr)) | (d$thickness > (q3 + 1.5*iqr))
d$thickness[out_iqr & !is.na(d$thickness)] <- NA
clean_outlier <- d箱线图查看处理后分布
boxplot(clean_outlier$age, clean_outlier$thickness, names = c("age", "thickness"), main = "异常值处理后的分布")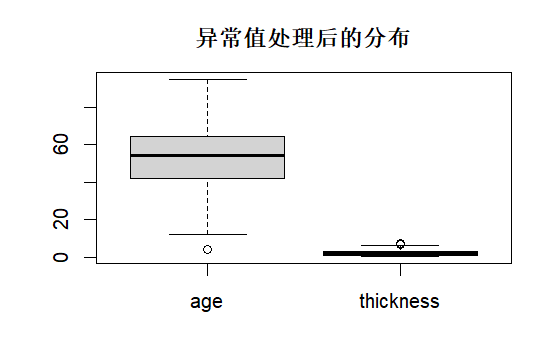
(3)重复数据处理
library(dplyr)
# 在 clean_outlier 基础上(若未做上一步则用 clean_missing 或 Melanoma_dirty)
d <- as.data.frame(clean_outlier)
# 按业务主键去重:time + age + thickness + sex 视为一条记录身份,保留第一条
clean_dup <- d %>%
arrange(time, age, thickness, sex) %>%
distinct(time, age, thickness, sex, .keep_all = TRUE)
cat("去重前行数:", nrow(d), " 去重后:", nrow(clean_dup), "\n")(4)数据标准化
# 在 clean_dup 基础上:统一 sex 编码("0"/"1"/"男"/"女" -> 统一为 "男"/"女")
d <- as.data.frame(clean_dup)
d$sex <- dplyr::case_when(
d$sex %in% c("1", "男", "M", "Male") ~ "男",
d$sex %in% c("0", "女", "F", "Female") ~ "女",
TRUE ~ NA_character_
)
clean_std <- d
# 验证:sex 仅剩两种取值
table(clean_std$sex, useNA = "ifany")3.2 数据集成与转换
在多源、多表的医学数据环境中,数据集成与转换是从“原始数据”走向“可分析数据集”的关键一步。
- 多源数据融合通过关键字匹配、规则匹配或机器学习匹配,将 HIS、LIS、PACS、随访系统等不同来源的数据对齐到同一患者/同一次就诊。
- 数据转换对变量进行标准化(Z-score)、归一化(Min-Max)、对数变换、离散化,以便模型训练或结果解释。
- 特征工程围绕研究问题进行特征提取、特征选择、特征构造,以及降维(如 PCA/FA)以压缩冗余信息。
以下均基于前文清洗结果 clean_std(若未做 3.1 则用 Melanoma_dirty 或 Melanoma),每个知识点一段完整代码。
(1)多源数据融合
拆成“基础信息表”与“肿瘤/结局表”
library(dplyr)
tbl_base <- clean_std %>% select(time, age, sex) %>% mutate(id = row_number())
tbl_outcome <- clean_std %>% select(time, thickness, ulcer, status) %>% mutate(id = row_number())按主键 id 合并
merged <- tbl_base %>%
left_join(tbl_outcome, by = "id") %>%
select(-ends_with(".y"))
head(merged)(2)数据转换
Z-score 与 Min-Max 归一化
d <- as.data.frame(clean_std)
d$age_z <- as.numeric(scale(d$age))
d$thickness_z <- as.numeric(scale(d$thickness))
d$age_norm <- (d$age - min(d$age, na.rm = TRUE)) / (max(d$age, na.rm = TRUE) - min(d$age, na.rm = TRUE))
d$thickness_norm <- (d$thickness - min(d$thickness, na.rm = TRUE)) /
(max(d$thickness, na.rm = TRUE) - min(d$thickness, na.rm = TRUE) + 1e-8)对数变换与离散化(年龄组、厚度分组)
d$log_time <- log1p(d$time)
d$log_thickness <- log1p(d$thickness)
d$age_grp <- cut(d$age, breaks = c(0, 40, 60, 100), labels = c("<40", "40-60", ">60"), include.lowest = TRUE)
d$thickness_cat <- cut(d$thickness, breaks = c(0, 1, 4, 10), labels = c("薄", "中", "厚"), include.lowest = TRUE)
transformed <- d查看转换结果
head(transformed[, c("age", "age_z", "age_norm", "age_grp", "log_thickness", "thickness_cat")])(3)特征工程
特征构造:年龄×厚度、是否高龄
d <- as.data.frame(clean_std)
d$age_thick <- d$age * d$thickness
d$is_older <- (d$age > 60) + 0PCA 降维:取前 2 个主成分
num_cols <- c("time", "age", "thickness", "ulcer", "year")
X <- scale(d[, num_cols], center = TRUE, scale = TRUE)
X <- X[complete.cases(X), ]
pc <- prcomp(X)
summary(pc)
feat_pc <- pc$x[, 1:2]
colnames(feat_pc) <- c("PC1", "PC2")
head(feat_pc)四、挑战与伦理
医学数据分析与数据挖掘在带来价值的同时,也面临技术难点与伦理、法规约束。
4.1 技术挑战
- 数据质量缺失、噪声、异构性(多源、多格式、多标准),影响模型稳健性与结论可靠性
- 隐私保护患者敏感信息安全、数据匿名化与脱敏,在可用性与隐私之间取得平衡
- 可解释性黑箱模型(如复杂深度学习)带来的临床信任问题,需可解释 AI 与临床可接受性
- 标准化多中心数据整合、术语与编码统一(如 ICD、SNOMED),实现跨机构可比与复用
4.2 伦理与法规
- 知情同意数据使用授权、研究目的告知、隐私保护承诺,确保患者知情与同意
- 数据安全存储加密、传输安全、访问控制与权限管理,防止泄露与滥用
- 法规遵循HIPAA(美国)、GDPR(欧盟)及国内医疗数据管理规范,合规使用与跨境注意适用法律